Tesamorelin Peptide: Structure, Mechanism, and Research Applications
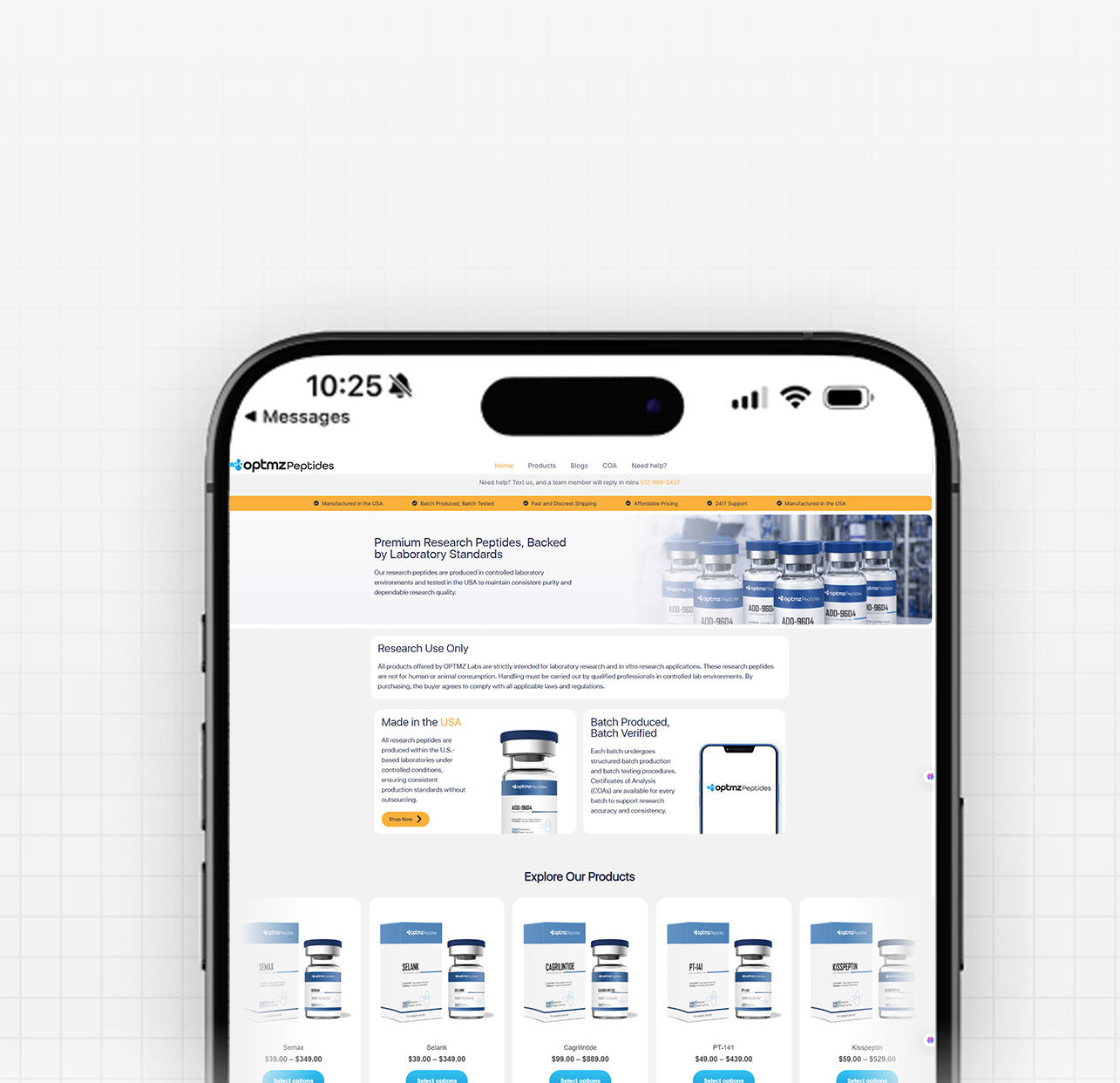
By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published April 18, 2026 · Last updated April 18, 2026 Tesamorelin is a synthetic 44-amino-acid analog of human growth hormone-releasing hormone (GHRH), originally developed under the code TH9507. It is chemically designated N-(trans-3-hexenoyl)-[Tyr¹]hGRF(1-44)NH₂ acetate — a GHRH(1-44) sequence modified at the N-terminal tyrosine with a hexenoyl fatty acid group that resists proteolytic cleavage. This modification is the central reason tesamorelin has been studied as a stabilized pharmacological probe for GHRH receptor signaling in research contexts (Ferdinandi et al., 2007, PMID 17214611). This page summarizes the molecular structure, receptor pharmacology, published research literature, and analytical verification standards relevant to tesamorelin as a research-grade peptide. All references are cited from peer-reviewed sources. Mechanistic claims are anchored to PubMed-indexed studies. What Is Tesamorelin? Tesamorelin is a synthetic growth hormone-releasing factor (GRF) analog derived from the full 44-amino-acid sequence of human GHRH (hGRF). Unlike shorter GHRH-pathway peptides such as sermorelin (GHRH 1-29), tesamorelin retains the complete native sequence and adds a structural modification at the N-terminus that prevents rapid enzymatic degradation. The compound activates the GHRH receptor (GHRH-R), a G protein-coupled receptor (GPCR) expressed on pituitary somatotropes. Receptor binding drives pulsatile release of endogenous growth hormone (GH) rather than introducing exogenous GH directly — a pharmacological distinction that has made it an object of research interest in studies of the GH/IGF-1 axis (Wang, 2009, PMID 19243281). The Molecular Structure of Tesamorelin Tesamorelin’s full chemical designation is N-(trans-3-hexenoyl)-[Tyr¹]hGRF(1-44)NH₂ acetate. Breaking this down: hGRF(1-44)NH₂ — the 44-amino-acid sequence of human growth hormone-releasing factor, C-terminally amidated [Tyr¹] — tyrosine at position 1, retained from native GHRH N-(trans-3-hexenoyl) — a six-carbon unsaturated fatty acid (hexenoyl) group covalently attached to the N-terminus acetate — the salt form used in pharmaceutical and research-grade preparations The trans-3-hexenoyl modification is the functional innovation. Native GHRH is cleaved rapidly by the enzyme dipeptidyl peptidase-IV (DPP-IV) at the Tyr¹-Ala² bond, producing the inactive GHRH(3-44) metabolite within minutes of administration. Attaching the hexenoyl group to the N-terminal tyrosine sterically blocks DPP-IV access, extending the molecule’s functional half-life in plasma (Ferdinandi et al., 2007). The molecular weight of tesamorelin acetate is approximately 5,196 Da, which places it in the mid-range for synthetic peptide research compounds and informs HPLC method selection for purity verification. How Does Tesamorelin Interact With the GHRH Receptor? Tesamorelin binds the GHRH receptor (GHRH-R), a class B GPCR located primarily on somatotroph cells in the anterior pituitary. The signal cascade has been characterized as follows in published research: Receptor binding: Tesamorelin engages the extracellular N-terminal domain and transmembrane core of GHRH-R, mimicking the binding pose of native GHRH. Gαs activation: Conformational change activates the heterotrimeric Gs protein, stimulating adenylate cyclase. cAMP elevation: Intracellular cAMP rises, activating protein kinase A (PKA) and CREB-mediated transcription of the GH gene. Pulsatile GH release: Somatotropes release stored GH into circulation in discrete pulses — a pattern that preserves the native pulsatility of endogenous GH secretion rather than producing sustained hormone elevation. Hepatic IGF-1 induction: Circulating GH binds hepatic GH receptors, driving synthesis and release of insulin-like growth factor-1 (IGF-1), the major downstream effector of the GH axis. Stanley and colleagues observed that tesamorelin administration in a controlled research population preserved physiological pulsatile GH secretion patterns and did not produce the insulin resistance commonly reported with exogenous GH administration (Stanley et al., 2011, PMID 20943777). This finding is frequently cited as the mechanistic rationale for classifying tesamorelin as a GHRH secretagogue rather than a GH replacement analog. What Has Research Examined About Tesamorelin? Published tesamorelin research spans pharmacokinetic characterization, GH-axis modulation, and metabolic parameter analysis. The following areas are well-represented in the peer-reviewed literature: GH and IGF-1 Axis Modulation Multiple controlled studies have measured circulating GH and IGF-1 changes following tesamorelin administration. Stanley et al. (2011) documented preserved pulsatile GH secretion and a rise in mean IGF-1 levels without evidence of the supraphysiological IGF-1 surges associated with recombinant GH protocols. Pharmacokinetics and Metabolic Parameters Clemmons and colleagues reported 12-month data on tesamorelin pharmacokinetics and metabolic parameters (including glucose tolerance markers) in a controlled research population (Clemmons et al., 2017, PMC5472315). The dataset remains one of the longest continuous research observations of GHRH-pathway modulation published in the analog class. Clinical Literature on HIV-Associated Lipodystrophy The majority of published tesamorelin human research has been conducted in the context of HIV-associated lipodystrophy, a condition characterized by altered regional adipose distribution. Spooner and Olin (2012, PMID 22298602) reviewed the clinical trial literature in this population, and a 2024 meta-analysis examined randomized controlled trial outcomes across the literature base (Badran et al., 2024). OPTMZ Peptides reports this research record as documentary reference — we do not promote tesamorelin as a consumer product for any indication. Broader Metabolic Research Recent review literature has situated tesamorelin within the larger context of GHRH-pathway research in diabetes and metabolic physiology (Steenblock & Bornstein, 2024, PMC12137473). Ongoing research continues to characterize GHRH receptor distribution in peripheral tissues and the role of local GHRH signaling outside the pituitary. Tesamorelin vs. Native GHRH: Pharmacokinetic Differences Parameter Native GHRH Tesamorelin Amino acid length 44 (hGRF 1-44) 44 (hGRF 1-44) N-terminal modification None trans-3-hexenoyl group DPP-IV resistance No — cleaved at Tyr¹-Ala² Yes — hexenoyl blocks access Plasma half-life ~6-7 minutes ~26-38 minutes (published range) Receptor affinity Reference Comparable to native GHRH The extended half-life is the primary pharmacological rationale for tesamorelin’s development. Native GHRH’s ~7-minute half-life makes it impractical as a research probe for studies requiring sustained receptor engagement; tesamorelin’s 4–5x extended window enables controlled experimental designs that would be logistically impossible with native GHRH (Ferdinandi et al., 2007). Tesamorelin vs. Sermorelin and Other GHRH-Pathway Peptides Researchers frequently compare tesamorelin with other peptides that engage the GHRH pathway. The key structural and pharmacological distinctions: Sermorelin (GHRH 1-29 NH₂) Shortened 29-amino-acid fragment of GHRH retaining the biologically active N-terminal region No DPP-IV resistance modification — rapid plasma clearance Generally used in research protocols requiring shorter receptor engagement windows CJC-1295 (DAC) Modified GHRH(1-29) conjugated to a drug affinity complex (DAC) that binds serum
BPC-157, Nitric Oxide, and Angiogenesis: A Research Summary of the VEGFR2–NO Pathway

BPC-157 is a synthetic peptide that has been widely studied for its role in tissue repair and cellular signaling. One area that continues to draw attention is its potential interaction with nitric oxide, a molecule involved in blood flow and vascular function. Understanding this relationship helps researchers explore how peptides may influence key biological systems. What Is BPC-157? BPC-157 is a peptide derived from a protein found in gastric juice. In research settings, it is often studied for its connection to healing processes, including how tissues respond to stress or injury. Its stability and ability to interact with multiple pathways make it a frequent subject in peptide research. What Is Nitric Oxide? Nitric oxide is a signaling molecule produced naturally in the body. It plays a major role in regulating blood vessel function by helping vessels relax and expand. This process, known as vasodilation, allows for better blood flow and nutrient delivery throughout the body. How BPC-157 and Nitric Oxide May Be Connected Research suggests that BPC-157 may interact with pathways related to nitric oxide production and regulation. Some studies indicate that it may: Influence nitric oxide signaling pathways Support vascular response mechanisms Affect blood flow at the tissue level Interact with processes involved in repair and regeneration These interactions are of interest because nitric oxide is closely tied to how the body responds to injury and maintains circulation. Areas of Research Interest Researchers explore this connection across several areas: Vascular FunctionStudying how nitric oxide pathways affect blood flow and how peptides may influence those responses. Tissue Repair ProcessesUnderstanding how improved circulation and signaling contribute to healing at the cellular level. Cellular Signaling MechanismsAnalyzing how different pathways interact and regulate biological responses. What Current Research Suggests Most findings come from preclinical and laboratory studies. These suggest that BPC-157 may interact with nitric oxide systems in ways that influence vascular and cellular responses. However, results vary depending on the model and conditions used in research. Limitations and Ongoing Study Here’s the honest picture. While there is growing interest in the connection between BPC-157 and nitric oxide, large-scale human research is still limited. Most available data comes from: Animal studies Cell-based experiments Controlled laboratory research More research is needed to fully understand how these interactions behave across different contexts. Final Thoughts BPC-157 and nitric oxide are both important in understanding how the body regulates blood flow and tissue response. Their potential interaction makes them a valuable focus in ongoing peptide research. At this stage, this area remains under active investigation, with future studies expected to provide deeper insights into how these systems work together.
What Is Cagrilintide? A Research Overview of the Long-Acting Amylin Analog

Cagrilintide is a synthetic peptide analog that has gained attention in research for its interaction with pathways related to appetite regulation and metabolic signaling. It is designed to mimic the activity of a naturally occurring hormone called amylin. Because of this, it’s often studied in connection with how the body manages energy balance and food intake. What Is Cagrilintide? Cagrilintide is an analog of amylin, a hormone that is normally released alongside insulin. Amylin plays a role in regulating appetite, slowing gastric emptying, and influencing how the body processes nutrients. By mimicking this hormone, Cagrilintide is studied to understand how these same pathways can be influenced in a controlled research setting. How It Works In research, Cagrilintide is examined for how it interacts with receptors involved in appetite and metabolic control. Some of its studied effects include: Influencing satiety signals Slowing the rate at which food leaves the stomach Affecting energy intake behavior Interacting with metabolic regulation pathways Rather than acting directly on energy production, it works through signaling mechanisms that help regulate intake and processing. Why It’s Being Studied Cagrilintide has become a focus in research because it targets specific pathways related to metabolic balance. Researchers are particularly interested in: Appetite RegulationUnderstanding how signals in the brain and body control hunger and fullness. Energy BalanceStudying how intake and expenditure are regulated through hormonal signaling. Peptide-Based PathwaysExploring how synthetic analogs can replicate and modify natural hormone activity. What Research Suggests So Far Most findings come from controlled studies and early-stage research. These suggest that Cagrilintide can influence appetite-related pathways through its interaction with amylin receptors. However, outcomes can vary depending on study design, dosage, and experimental conditions. Limitations and Current Understanding Here’s the honest take. While research is ongoing, there is still limited long-term data available across broader populations. Much of the current understanding is based on: Early-stage clinical research Laboratory studies Controlled experimental settings Further research is needed to fully understand its long-term behavior and broader implications. Final Thoughts Cagrilintide is a synthetic peptide designed to mimic amylin, making it a key subject in research focused on appetite and metabolic signaling. Its targeted mechanism has made it an area of growing interest. At this stage, it remains an active field of study, with ongoing research continuing to explore its full potential and applications.
GHK-Cu Peptide: Research Overview, Mechanism, and Batch Verification
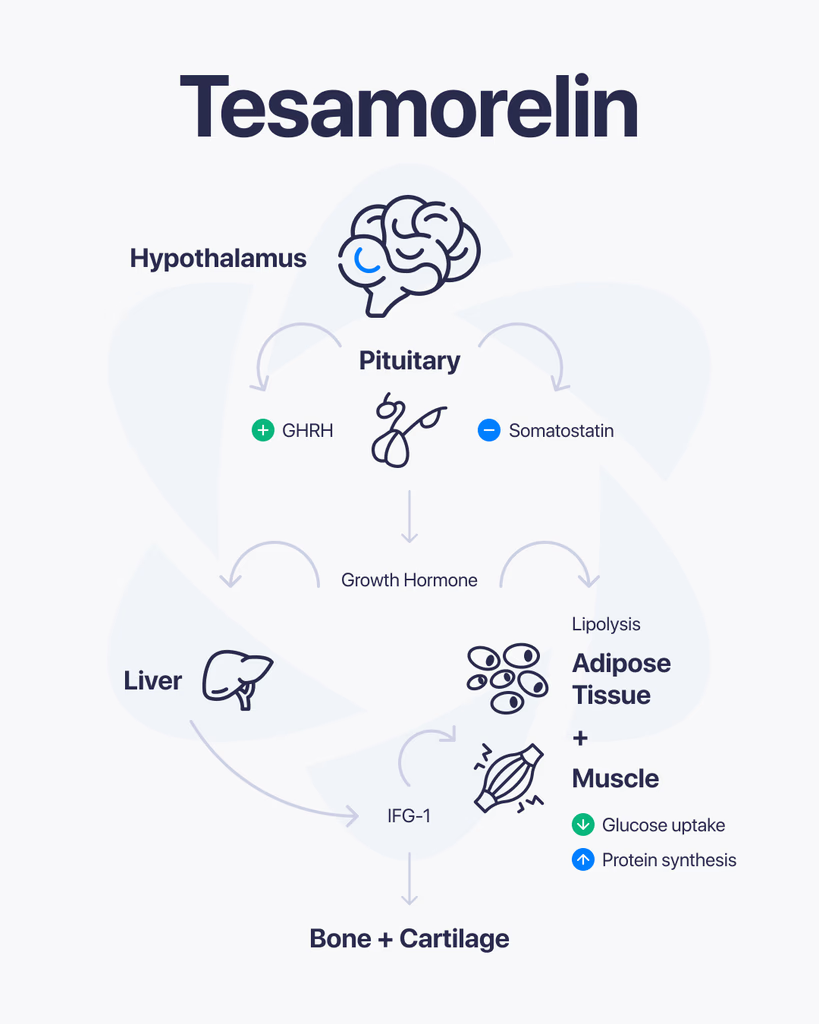
GHK-Cu is a naturally occurring peptide complex that has been widely studied for its role in cellular activity and tissue-related processes. It consists of a short peptide (GHK) bound to copper ions, which gives it unique properties in research settings. Because of this combination, it has become a point of interest in studies focused on cellular repair, signaling, and biological balance. What Is GHK-Cu Made Of? GHK-Cu is formed from a tripeptide made up of three amino acids: Glycine Histidine Lysine When this peptide binds with copper, it creates a complex that can interact with various biological systems. The copper component plays a key role in how the compound functions at the cellular level. How It Works in Research In research environments, GHK-Cu is studied for how it influences cellular communication and tissue-related processes. Some areas of focus include: Cellular repair mechanisms Regulation of gene expression Interaction with enzymes and proteins Support of structural components in tissues Its ability to bind copper allows it to participate in processes where metal ions are involved in biological activity. Why It’s Studied GHK-Cu has drawn attention because of its potential to influence multiple pathways at once. Researchers are interested in how it may support: Tissue-Related ProcessesStudying how cells repair and maintain structure. Cellular SignalingUnderstanding how signals are transmitted between cells. Structural Protein InteractionAnalyzing how it may affect components like collagen and other proteins. What Research Suggests Most available research comes from laboratory and preclinical studies. These suggest that GHK-Cu may play a role in regulating cellular processes and supporting tissue-related functions. However, outcomes can vary depending on conditions such as concentration, environment, and study design. Limitations and Current Understanding Here’s the honest picture. While GHK-Cu has been studied for many years, there is still ongoing research to fully understand all of its mechanisms and long-term effects. Most findings are based on: Cell-based studies Animal research Controlled laboratory environments More extensive research is needed to expand current knowledge. Final Thoughts GHK-Cu is a well-known peptide complex in research due to its connection to copper and its involvement in cellular processes. Its ability to interact with multiple biological pathways makes it an important subject of ongoing study. At this stage, it remains a key area of interest in peptide research, with continued investigation helping to uncover its full range of activity.
AOD-9604 Peptide: A Research Overview of Mechanisms, Studies, and Verification Standards
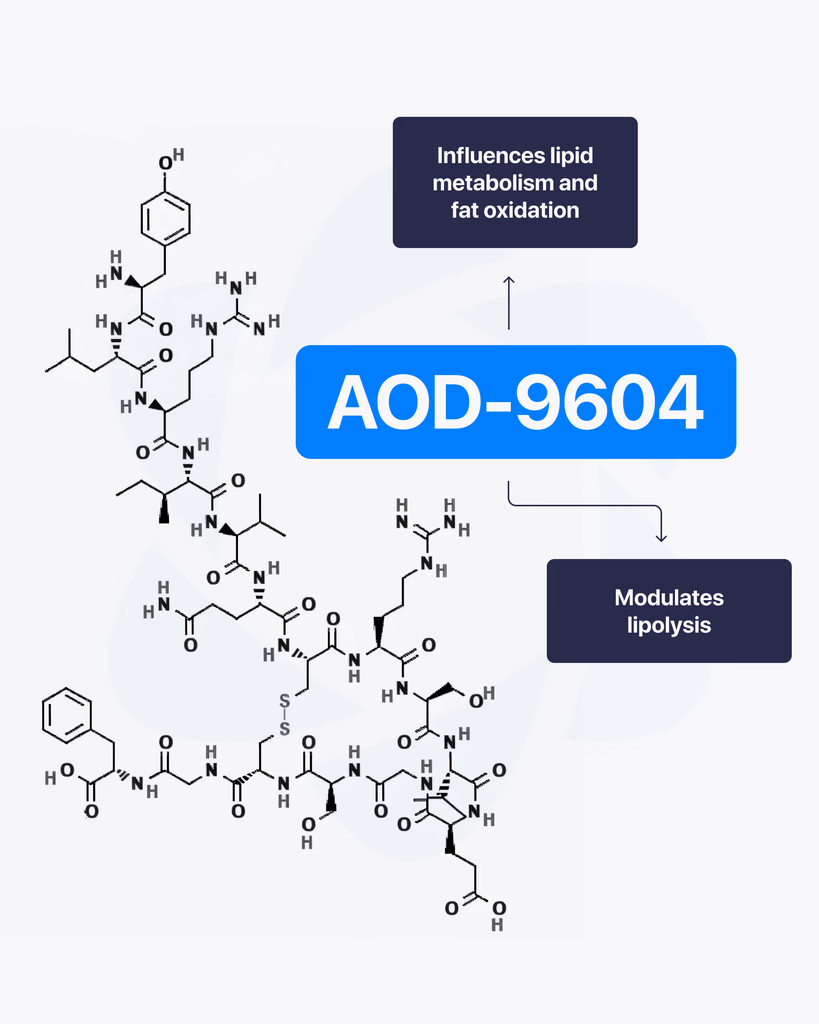
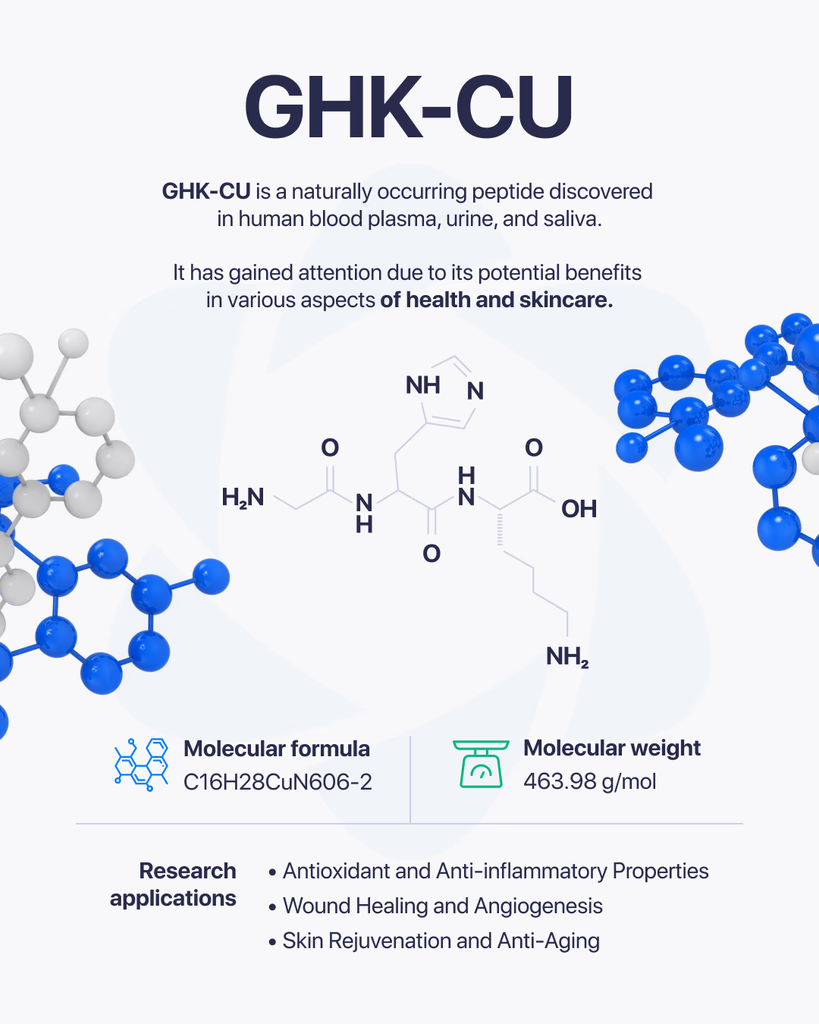
By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published April 2, 2026 | Last updated April 16, 2026 Research Use Only. All products and information referenced in this article are intended for laboratory research conducted by qualified professionals. AOD-9604 is not approved by the FDA for any clinical use and is not intended for human or animal consumption. Statements in this article have not been evaluated by the FDA and are not intended to diagnose, treat, cure, or prevent any disease. AOD-9604 is a synthetic 16-amino-acid peptide corresponding to the C-terminal fragment (residues 176–191) of human growth hormone (hGH). It was developed in the late 1990s by Metabolic Pharmaceuticals (Australia) and has been investigated in pre-clinical metabolic research and, more recently, in tissue and cartilage research models. AOD-9604 is sold for laboratory research use only; it is not an FDA-approved therapeutic agent. What Is AOD-9604? AOD-9604 is a hexadecapeptide engineered to replicate a specific functional segment of hGH — the lipolytic domain — without reproducing the full hormone’s growth-promoting or insulin-modulating activity. Researchers refer to the same molecule by several names: AOD-9604, AOD9604, hGH fragment 176-191, and (less commonly in current literature) hGH 177-191. The compound carries CAS Number 221231-10-3, a molecular formula of C₇₈H₁₂₃N₂₃O₂₃S₂, and a molecular weight of approximately 1,815.1 g/mol. Its PubChem CID is 71300630. The molecule was synthesized to investigate whether the lipid-pathway-related activity of growth hormone could be isolated from its other endocrine effects. In animal model research conducted at Monash University and by Metabolic Pharmaceuticals, AOD-9604 was reported to induce changes in lipid metabolism without producing the IGF-1 elevation, glucose effects, or growth signaling associated with full-length hGH (Ng et al., 2000) PubMed: 11146367. AOD-9604 Molecular Structure and Origin: The HGH Fragment 176-191 The full hGH molecule contains 191 amino acids. Early structure-activity research conducted in the 1980s and 1990s identified the C-terminal region — specifically residues 177–191, with a tyrosine added at position 176 to produce a stable hexadecapeptide — as the segment most associated with measurable lipid-pathway effects in vitro and in animal models. Metabolic Pharmaceuticals patented the resulting analogue, designated AOD-9604, in the late 1990s and advanced it through a series of pre-clinical and Phase I/II investigations between 2001 and 2007 (Stier et al., 2013) jofem.org. A 2010 review of obesity pharmacology characterized AOD-9604 as an orally and parenterally bioavailable analogue of the hGH 177-191 region that demonstrated lipolytic activity in animal trials but produced limited efficacy signals in later-stage human studies (Isidro & Cordido, 2010). The compound’s clinical development as a therapeutic was discontinued in 2007 following Phase 2B trial outcomes that did not support commercial advancement (Valentino et al., 2010) PMC3136748. For research-grade verification, the molecular structure is commonly confirmed by mass spectrometry (target m/z corresponding to the 1,815.1 g/mol molecular weight) and purity is quantified by reversed-phase HPLC against a reference standard. These methods, along with endotoxin and microbial testing, comprise the standard analytical panel applied to AOD-9604 batches sold for research use. How Does AOD-9604 Work in Research Models? The most-studied biochemical activity of AOD-9604 in pre-clinical models involves interaction with lipid-metabolism pathways in adipose tissue. Heffernan and colleagues, working at Monash University, examined chronic AOD-9604 administration in obese mice and in β3-adrenergic receptor knock-out mice. Their 2001 paper in Endocrinology reported that the compound produced effects on lipolytic sensitivity that appeared to depend at least partially on β3-adrenergic receptor signaling — a pathway distinct from the GH receptor — and did not measurably stimulate IGF-1 production (Heffernan et al., 2001) PubMed: 11713213. A separate study by Ng and colleagues in Hormone Research examined AOD-9604 in obese Zucker rats, a standard rodent model in metabolic research, and reported observations consistent with isolated lipid-pathway activity in the absence of measurable somatogenic effects (Ng et al., 2000) PubMed: 11146367. Together, these foundational papers established the research framework in which AOD-9604 has subsequently been investigated. It is important to note that the mechanism described in animal models has not translated into clinically meaningful human efficacy. The Phase 2B human trial conducted by Metabolic Pharmaceuticals between 2004 and 2007 did not produce results sufficient to support continued development, and AOD-9604 has not been approved by the FDA, EMA, or TGA for any therapeutic indication. What Have Published Studies of AOD-9604 Examined? The published literature on AOD-9604 spans roughly 25 years and includes pre-clinical animal studies, human safety trials, and more recent investigations in tissue research contexts. Pre-clinical metabolic research The earliest characterizations of AOD-9604 focused on lipid-pathway interactions in rodent models. Ng et al. (2000) and Heffernan et al. (2001) remain the most-cited references for the foundational pharmacology, and both papers are openly accessible through PubMed. Human safety and tolerability research Stier and colleagues published a 2013 review in the Journal of Endocrinology, Metabolism and Diabetes of South Africa summarizing six human clinical trials conducted between 2001 and 2006 involving 893 participants. The review reported that AOD-9604 was generally well-tolerated in short-term administration and did not produce clinically significant changes in IGF-1, glucose, or insulin parameters compared to placebo (Stier et al., 2013) jofem.org. A subsequent 2014 publication by Moré and colleagues reviewed long-term safety and genotoxicology data and reported no evidence of mutagenic or toxicological concerns within the parameters studied (Moré et al., 2014). Cartilage and tissue research More recent literature has examined AOD-9604 in the context of cartilage and connective tissue research. A 2026 review of therapeutic peptides in orthopaedic research models discussed AOD-9604 alongside other peptide fragments studied for their potential interactions with chondrocyte signaling and tissue maintenance pathways (Rahman et al., 2026) PMC12753158. This research direction remains preliminary, with most published work confined to in vitro and small-animal models. Across all three research contexts, the published data is best characterized as exploratory. Sample sizes are limited, the body of human evidence is small, and no large-scale efficacy trials have been completed for any indication. What Is AOD-9604’s Regulatory Status? AOD-9604 is not approved by the U.S. Food and Drug Administration for any clinical use. It
NAD+ Peptide Research: Mechanisms, Studies & Verification
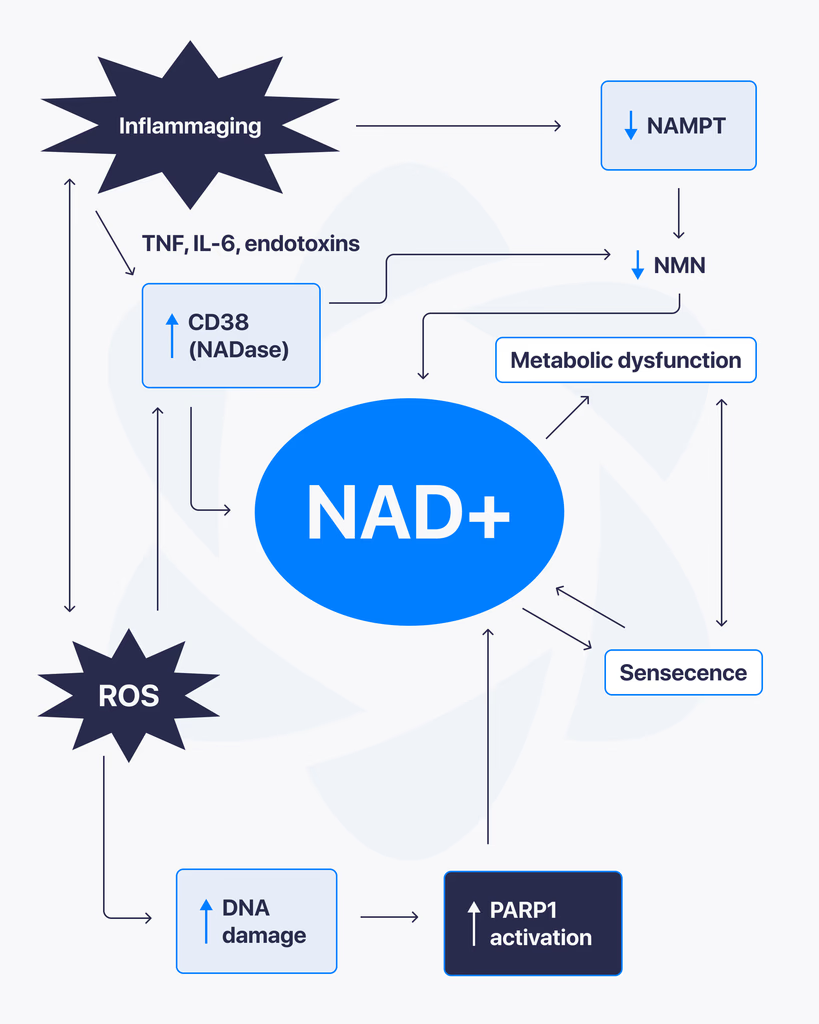
NAD+, short for nicotinamide adenine dinucleotide, is a coenzyme found in every living cell. It plays a central role in energy production and cellular function, which is why it has become a major focus in ongoing research. Scientists are studying how NAD+ levels influence different biological processes, especially those related to metabolism and cellular health. What Is NAD+? NAD+ is a molecule that helps transfer energy within cells. It’s involved in reactions that convert nutrients into usable cellular energy. As cells perform their functions, NAD+ cycles between two forms, helping drive essential processes that keep cells active and functioning. Why NAD+ Matters NAD+ is critical for several core cellular functions, including: Energy production within mitochondria DNA repair mechanisms Regulation of cellular stress responses Support of metabolic processes Because of its wide-ranging role, changes in NAD+ levels can affect how efficiently cells operate. NAD+ and Cellular Energy One of the main reasons NAD+ is studied is its connection to energy metabolism. It helps facilitate reactions that produce ATP, the primary energy source used by cells. Without sufficient NAD+, these processes become less efficient, which can impact overall cellular performance. What Research Is Exploring Researchers are examining how NAD+ interacts with different pathways inside the body. Some key areas of focus include: Mitochondrial FunctionUnderstanding how NAD+ supports energy production and efficiency within cells. Cellular Repair SystemsStudying its role in activating enzymes involved in DNA repair and maintenance. Metabolic RegulationExploring how NAD+ levels influence how the body processes and uses energy. What Studies Suggest So Far Most findings come from laboratory and preclinical research. These studies indicate that NAD+ plays a vital role in maintaining cellular function and responding to stress. There is also growing interest in how NAD+ levels change over time and how that may affect different biological systems. Limitations and Current Understanding Here’s the reality. While NAD+ is well understood at a biochemical level, many of its broader effects are still being researched. Most available data comes from: Cell-based studies Animal research Early-stage human investigations More comprehensive research is needed to fully understand long-term impacts and practical applications. Final Thoughts NAD+ is a key molecule in cellular energy and maintenance, making it an important subject in modern research. Its involvement in multiple biological pathways continues to drive interest across different scientific fields. At this stage, it remains an active area of study, with ongoing research working to better understand how it influences cellular function over time.
How Does Sermorelin Work? GHRH Mechanism & Research Overview

By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published: April 2, 2026 · Last updated: April 16, 2026 Sermorelin is a 29-amino-acid synthetic peptide corresponding to residues 1–29 of growth hormone-releasing hormone (GHRH 1–44), the biologically active N-terminal fragment. In research models, sermorelin binds the GHRH receptor on anterior pituitary somatotrophs, activates adenylate cyclase, and triggers a cyclic AMP–protein kinase A (cAMP–PKA) signaling cascade that has been studied for its role in growth hormone gene transcription and release (Walker, 2006). Investigators have used sermorelin to examine GHRH-receptor pharmacology, downstream second-messenger dynamics, and — more recently — its interactions with cell proliferation pathways in oncology research models (Chang et al., 2021). This research overview summarizes what controlled studies have documented about sermorelin’s molecular mechanism, its half-life behavior in research contexts, what cell proliferation data has emerged, and how research-grade material is verified for laboratory use. What Is Sermorelin? Sermorelin is the truncated 1–29 amino acid sequence of native human GHRH, retaining the receptor-binding domain required for GHRH-R activation. The full-length endogenous peptide is 44 amino acids, but research has established that the first 29 residues are sufficient to reproduce the agonist activity of the parent molecule (Walker, 2006). The compound is supplied for research as a lyophilized powder requiring reconstitution in bacteriostatic water before use in cell-based or in-vivo research protocols. Structurally, sermorelin is classified as a GHRH analog rather than a growth hormone analog — a distinction that matters for experimental design. It does not interact directly with growth hormone receptors. Instead, it acts upstream at the pituitary level, making it a tool for studying GHRH-R signaling rather than peripheral GH-receptor activity. How Does Sermorelin Work? The GHRH Receptor Mechanism The mechanism of action observed in pre-clinical research follows a defined sequence at the molecular level: Receptor binding. Sermorelin binds to the GHRH receptor (GHRH-R), a class B G-protein-coupled receptor (GPCR) expressed on somatotroph cells of the anterior pituitary. G-protein coupling. Receptor activation triggers conformational changes that activate the Gαs subunit of the associated heterotrimeric G-protein. Adenylate cyclase activation. Gαs stimulates membrane-bound adenylate cyclase, which catalyzes the conversion of ATP to cyclic AMP (cAMP). PKA cascade. Elevated intracellular cAMP activates protein kinase A (PKA), which phosphorylates target proteins including the transcription factor CREB. Transcriptional regulation. Phosphorylated CREB binds the GH gene promoter, increasing transcription of growth hormone messenger RNA. Walker (2006) noted that this pathway “stimulates pituitary gene transcription of hGH messenger RNA, increasing pituitary reserve.” This signaling architecture is what makes sermorelin useful in research: it engages a well-characterized GPCR pathway with measurable second-messenger readouts (cAMP accumulation assays, CREB phosphorylation Western blots, GH ELISA from culture supernatant). A practical consequence observed in research contexts is that GH release driven by sermorelin remains under physiological feedback control by somatostatin and IGF-1, producing pulsatile rather than tonic patterns in animal models — a behavior distinct from exogenous recombinant GH administration (Walker, 2006). What Is the Half-Life of Sermorelin in Research Models? Sermorelin has a notably short plasma half-life — published research data places it at approximately 11–12 minutes in circulation, which is characteristic of GHRH-family peptides and reflects rapid enzymatic degradation by dipeptidyl peptidase-IV (DPP-IV) and other peptidases (Walker, 2006). For research workflows, this short half-life has several implications: Sampling windows are narrow. Time-course experiments measuring GH release require closely spaced sampling intervals (typically every 5–15 minutes for the first hour). Storage and reconstitution discipline matters. Once reconstituted, sermorelin should be maintained at 2–8°C and used within the verified stability window indicated on the batch certificate of analysis. Comparison studies use longer-acting analogs. Researchers comparing pulsatile versus sustained GHRH-R activation often pair sermorelin against modified GHRH analogs with extended half-lives (CJC-1295 and modified GRF 1–29 derivatives). What Has Research Shown About Sermorelin and Cell Proliferation? The most cited contemporary research on sermorelin and cellular proliferation is Chang et al. (2021), which used bioinformatic analysis (gene ontology and KEGG pathway enrichment) to examine sermorelin’s potential as a candidate for recurrent glioma research. The investigators reported that gene ontology analysis identified sermorelin as “closely related to cell proliferation functions” and proposed that the compound may inhibit tumor cell proliferation through cell cycle blocking mechanisms. Independent supporting work by Muñoz-Moreno et al. (2018) investigated GHRH-receptor antagonists in LNCaP and PC3 prostate cancer cell lines. While that work studied antagonists rather than agonists, it established that the GHRH-R signaling axis is functionally engaged in proliferative cell models — providing the receptor-pathway context that makes sermorelin’s effects in Chang’s bioinformatic analysis biologically plausible. It is important to characterize this body of work accurately: the cell proliferation data is bioinformatic and pre-clinical. There are no large-scale human studies investigating sermorelin’s effects on cellular proliferation. Researchers working in this area are typically running cell-line experiments, animal models, or computational analyses — not clinical trials. For laboratory teams designing experiments in this space, the key methodological considerations are: receptor expression validation in the chosen cell line (GHRH-R is not expressed at meaningful levels in all immortalized lines), batch-to-batch purity verification of the sermorelin used (impurities or degradation products can confound proliferation readouts), and inclusion of appropriate controls for cAMP-PKA pathway activation. How Does Sermorelin Compare to Other GHRH-Related Research Peptides? Within the GHRH-analog family, sermorelin is distinguished by its short half-life and its identity as the unmodified 1–29 sequence. Researchers comparing GHRH-pathway tools typically evaluate: CJC-1295 — a modified GRF 1–29 with a drug affinity complex (DAC) extension that significantly extends plasma half-life, used in research where sustained receptor engagement is the experimental variable. Tesamorelin — a stabilized GHRH 1–44 analog with an N-terminal trans-3-hexenoyl modification that resists DPP-IV cleavage. Ipamorelin — not a GHRH analog at all, but a ghrelin-receptor (GHS-R) agonist that converges on the same downstream output (GH release) through a parallel receptor system. This makes ipamorelin a useful comparator in studies dissecting the contributions of the two upstream pathways. The choice between these tools depends on the experimental question. Sermorelin’s short half-life and unmodified sequence make it the cleanest tool for studying acute,
MOTS-c Peptide: Mitochondrial-Derived Compound in Research
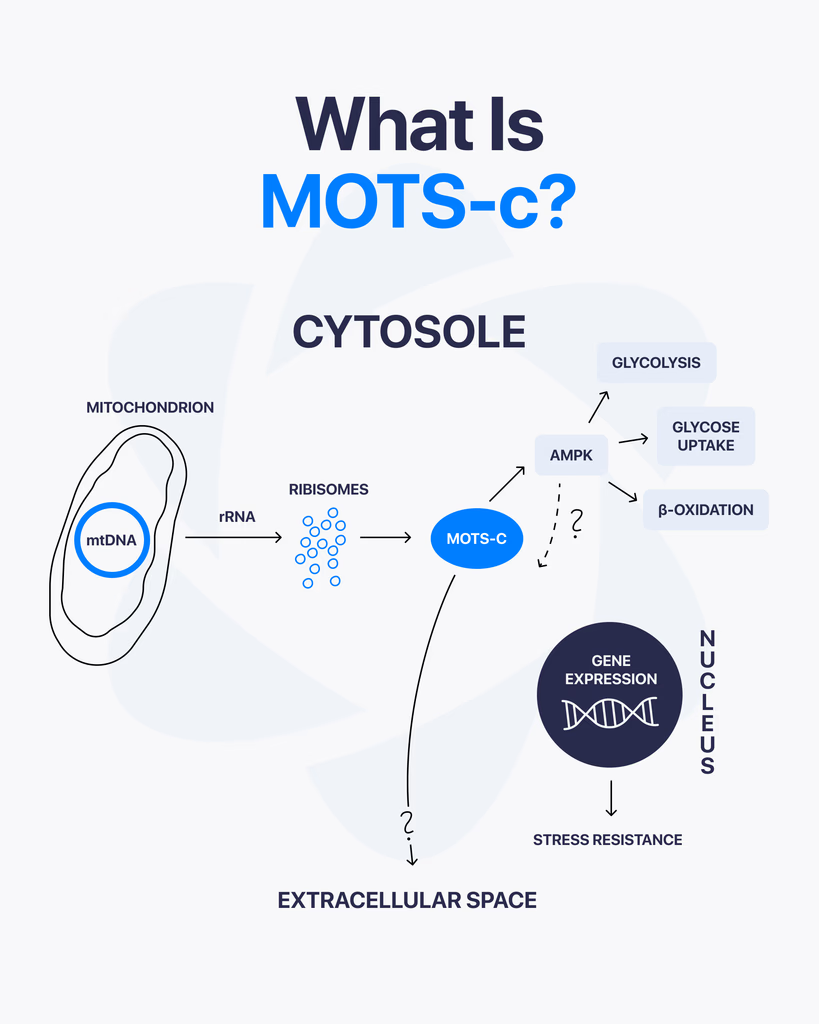
By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published April 2, 2026 · Last Updated April 16, 2026 MOTS-c is a 16-amino-acid mitochondrial-derived peptide encoded within the 12S rRNA region of mitochondrial DNA, and it has emerged as a focus of laboratory research into cellular metabolism and mitochondrial signaling. Discovered in 2015 by Lee and colleagues, MOTS-c is one of a small but growing class of peptides that originate inside the mitochondria rather than the cell nucleus, which is why it draws sustained attention in research focused on bioenergetics, exercise physiology models, and aging-related cellular signaling pathways. This page summarizes the published research literature on MOTS-c, the analytical specifications relevant to research-grade MOTS-c characterization, and the laboratory verification standards that apply to research peptide supply. All batch data referenced is independently verified by Krause Analytical, a DEA-registered, ISO/IEC 17025-certified laboratory in Austin, Texas. What Is MOTS-c? MOTS-c — Mitochondrial Open Reading Frame of the Twelve S rRNA-c — is a 16-amino-acid peptide with the sequence MRWQEMGYIFYPRKLR, encoded by a short open reading frame within the mitochondrial 12S rRNA gene. It belongs to a class of small mitochondrial-derived peptides (MDPs) that are translated from the mitochondrial genome rather than the nuclear genome (Lee et al., 2015 PMID: 25738459). Three structural features make MOTS-c notable in the published literature: Origin inside the mitochondrion. Unlike the majority of cellular peptides, which are encoded in nuclear DNA and trafficked to their site of action, MOTS-c is encoded directly within mitochondrial DNA. Short sequence with documented bioactivity in pre-clinical models. Research has examined MOTS-c interaction with metabolic regulatory pathways, including AMP-activated protein kinase (AMPK) signaling in skeletal muscle cell models (Lee et al., 2015). Detection in human plasma. Studies have measured circulating MOTS-c in human samples and reported its modulation by exercise in pre-clinical and translational research (Reynolds et al., 2021 PMID: 33473109). How Is MOTS-c Different from Other Research Peptides? Most research peptides supplied to laboratories — BPC-157, TB-500, GHK-Cu, and others — were either characterized from non-mitochondrial tissue or designed as synthetic analogues of nuclear-encoded signaling molecules. MOTS-c sits in a separate category: it is part of the mitochondrial-derived peptide (MDP) class, alongside humanin and the SHLP series. The structural distinction matters in research because mitochondrial-derived peptides have been examined as potential signaling molecules between the mitochondrion and other cellular compartments — a process referred to in the literature as mitochondrial-to-nuclear retrograde signaling (Kong et al., 2023). This positions MOTS-c as a research target in two distinct lines of inquiry: classical metabolic signaling research, and the emerging field of mitochondrial communication biology. What Does the Research Say About MOTS-c? Published research on MOTS-c is concentrated in pre-clinical and in vitro models. The major research areas covered in peer-reviewed literature include: Metabolic regulation in skeletal muscle models. The original Lee et al. (2015) paper reported that MOTS-c administration to murine models was associated with changes in glucose homeostasis and skeletal muscle glucose uptake, with a proposed mechanism involving AMPK pathway activation. Subsequent reviews have summarized this body of work and identified skeletal muscle as a primary research target tissue (Zheng et al., 2023 PMC9905433). Exercise-responsive expression. Reynolds and colleagues (2021) characterized MOTS-c expression patterns in response to exercise in murine and human samples, reporting that MOTS-c is responsive to acute exercise stimuli. The study examined MOTS-c expression in skeletal muscle and serum across multiple experimental conditions. Aging-related cellular signaling research. Review literature has examined MOTS-c in the context of aging-related cellular pathways, including pre-clinical investigations relevant to neurodegenerative, cardiovascular, and metabolic disease research models (Kong et al., 2023). It is important to note that this research is descriptive of laboratory observations, not therapeutic claims. Energy homeostasis in metabolic dysfunction models. A 2025 study by Pham and colleagues examined MOTS-c administration in laboratory models of metabolic dysfunction, reporting effects on energy homeostasis markers and muscle function in the experimental system (Pham et al., 2025). What current research does not establish: clinical efficacy in human populations, validated dosing protocols, long-term safety profiles, or any therapeutic application. Researchers reviewing this literature should treat all published data as descriptive of laboratory experimental systems. How Is MOTS-c Studied in Laboratory Research? Research-grade MOTS-c is typically supplied as a lyophilized powder produced by solid-phase peptide synthesis (SPPS), the standard methodology for laboratory peptide production. Laboratory protocols described in published research generally involve: Reconstitution in bacteriostatic or sterile water for in vitro experimental use Storage of lyophilized material at -20°C or below to preserve structural integrity Use within defined stability windows after reconstitution (typically 14–30 days at 2–8°C, depending on protocol and ambient handling conditions) HPLC and mass spectrometry verification of purity and identity prior to experimental use For research-grade MOTS-c sourced from suppliers, the most important verification step is review of the batch-specific Certificate of Analysis (COA). A complete COA documents purity by HPLC, identity confirmation by mass spectrometry, endotoxin levels, and the testing laboratory and date. What Are the Technical Specifications of Research-Grade MOTS-c? The following analytical specifications apply to research-grade MOTS-c characterization: Specification Value Amino acid sequence MRWQEMGYIFYPRKLR (16 residues) CAS Number 1627580-64-6 Molecular formula C₁₄₉H₂₆₀N₄₆O₄₆S₂ Molecular weight 2171.5 g/mol PubChem CID 91808068 Synthesis method Solid-phase peptide synthesis (SPPS) Form supplied Lyophilized powder Recommended storage (lyophilized) -20°C, protected from light Recommended storage (reconstituted) 2–8°C, use within 14–30 days Solubility Bacteriostatic water, sterile water Researchers requiring batch-specific analytical data — including the actual purity percentage, identity confirmation chromatogram, and endotoxin results for a specific lot — should consult the relevant Certificate of Analysis. OPTMZ publishes COAs for every batch in the Lab Results archive. How Is MOTS-c Purity Verified for Research Use? The minimum purity standard applied to research peptide supply varies by vendor. OPTMZ rejects any MOTS-c batch testing below 98% by HPLC; current batches typically test in the 98.5–99.9% range. Verification at the supplier level involves a defined panel of analytical tests: HPLC (high-performance liquid chromatography) quantifies the percentage of the target peptide relative to total peptide content. This is the primary purity measurement. Mass
KPV Peptide: Mechanism, Research Findings, and Verification Standards

By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published March 2, 2026 · Last Updated April 16, 2026 KPV is one of the most extensively researched short peptide fragments in the published literature on inflammatory signaling. First characterized as the C-terminal tripeptide of α-melanocyte-stimulating hormone (α-MSH), it has been the subject of cell-based and animal model research spanning intestinal, dermal, antimicrobial, and respiratory inflammation contexts since the early 2000s. A 2008 Gastroenterology publication by Dalmasso and colleagues remains one of the most-cited mechanistic studies on the peptide and is referenced more than 130 times in subsequent peer-reviewed work (Dalmasso et al., 2008 [PubMed]). For researchers evaluating KPV as a candidate compound for laboratory work, three questions tend to recur: what does the published mechanistic literature actually demonstrate, where are the limits of the current data, and how is research-grade KPV verified for purity and identity before it enters an experiment. This page addresses each in sequence. The structure that follows begins with the molecular and biochemical fundamentals — sequence, structure, derivation from α-MSH — then summarizes the principal areas of published research and the proposed mechanisms behind the observed effects. The page closes with the analytical methodology used to qualify research-grade KPV batches, including the seven-method panel run by Krause Analytical (DEA-registered, ISO/IEC 17025-certified) on every KPV batch supplied by OPTMZ Peptides. Each batch result is published in the OPTMZ Lab Analysis archive and is searchable by batch number from the vial label. All cited findings link to the original PubMed records. All purity claims correspond to specific, batch-level COA data — not to category-level marketing claims. What Is KPV Peptide? KPV is a tripeptide composed of three amino acids — lysine (K), proline (P), and valine (V) — that constitutes the C-terminal fragment of α-melanocyte-stimulating hormone (α-MSH). Research has examined KPV’s interaction with inflammatory signaling pathways in cell-based and animal models, with published studies investigating its activity in intestinal, dermal, and antimicrobial research contexts (Dalmasso et al., 2008 [PubMed]; Brzoska et al., 2008 [PubMed]). This page summarizes the current published research on KPV, its proposed mechanism of action, the experimental contexts in which it has been studied, and the analytical methods used to verify research-grade KPV batches for purity and identity. What Is the Molecular Structure of KPV? KPV is a linear tripeptide with the sequence Lys-Pro-Val. Its molecular formula is C₁₆H₃₀N₄O₄ and its molecular weight is approximately 342.4 Da. The three constituent amino acids are joined by two peptide bonds in the canonical N-to-C direction. KPV corresponds to residues 11–13 of α-MSH (the full sequence of α-MSH is Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH₂). This positioning at the C-terminus is significant: research has shown that the C-terminal fragment retains certain biological activities of the parent hormone while lacking the melanocortin receptor agonism associated with the full-length peptide (Brzoska et al., 2008 [PubMed]). Because of its small size, KPV has been investigated as a candidate for transdermal and oral delivery research, including iontophoretic delivery studies (Pawar et al., 2017 [PubMed]). How Is KPV Derived from α-MSH? α-MSH is a 13-residue peptide hormone produced by post-translational cleavage of pro-opiomelanocortin (POMC). Researchers have studied multiple α-MSH fragments — including KPV — for biological activity independent of the parent hormone’s receptor binding profile. KPV can be obtained either through enzymatic cleavage of α-MSH or through direct solid-phase peptide synthesis (SPPS). Research-grade KPV is typically produced via SPPS using Fmoc chemistry, followed by HPLC purification to research-grade purity standards (≥98%). What Does KPV Research Suggest About Inflammatory Pathways? Published research has investigated KPV’s interaction with several molecular components of inflammatory signaling. The most extensively cited body of work comes from intestinal inflammation models, where Dalmasso and colleagues demonstrated that KPV is taken up by intestinal epithelial cells via the PepT1 transporter and reduced markers of inflammation in murine colitis models (Dalmasso et al., 2008 [PubMed]). Subsequent investigations have examined KPV’s interaction with NF-κB signaling and with the production of pro-inflammatory cytokines including IL-6, IL-8, and TNF-α in cell-based assays. Research published in 2025 has additionally examined KPV’s effect on IL-1β production in models of fine-particulate-induced inflammation (Sung et al., 2025). It is important to note that all of these findings are derived from in vitro, ex vivo, and animal model research. Published clinical research in humans is limited. Which Research Areas Have Examined KPV? Intestinal Inflammation Models The 2008 Dalmasso study remains one of the most-cited investigations of KPV. Using both in vitro intestinal epithelial cell cultures and in vivo murine colitis models (DSS-induced and TNBS-induced), researchers reported reductions in inflammatory markers following KPV administration. The study identified PepT1-mediated cellular uptake as a proposed mechanism for KPV’s intracellular activity (Dalmasso et al., 2008 [PubMed]). Skin and Atopic Dermatitis Models KPV has been investigated in dermal research contexts, including iontophoretic transdermal delivery studies and topical formulation research. Pawar and colleagues characterized KPV’s permeation profile through skin models and reported that iontophoresis enhanced peptide delivery compared to passive diffusion (Pawar et al., 2017 [PubMed]). Antimicrobial Activity Studies Research on α-MSH-derived peptides has examined antimicrobial activity against several organisms, including Staphylococcus aureus and Candida albicans, with KPV identified as one of the active fragments retaining a portion of the parent peptide’s antimicrobial profile (Cutuli et al., 2000 [PubMed]; Brzoska et al., 2008 [PubMed]). Fine-Particulate Inflammation Models A 2025 publication examined KPV’s effect on inflammatory markers in cell models exposed to fine particulate matter, with researchers reporting modulation of IL-1β production via proposed antioxidant pathways (Sung et al., 2025). What Is the Reported Mechanism of Action? Across the published literature, three principal mechanisms have been proposed for KPV’s activity in research models: Cellular uptake via PepT1. Intestinal epithelial cells express the PepT1 di/tripeptide transporter, which Dalmasso and colleagues identified as the route by which KPV enters the cytoplasm. Once intracellular, KPV is proposed to interact with downstream signaling components rather than acting via cell-surface receptors (Dalmasso et al., 2008 [PubMed]). NF-κB pathway interaction. Multiple studies have reported that KPV exposure correlates with reduced activation of
Ipamorelin Peptide: A Research Review of Mechanism, Selectivity, and Purity Verification
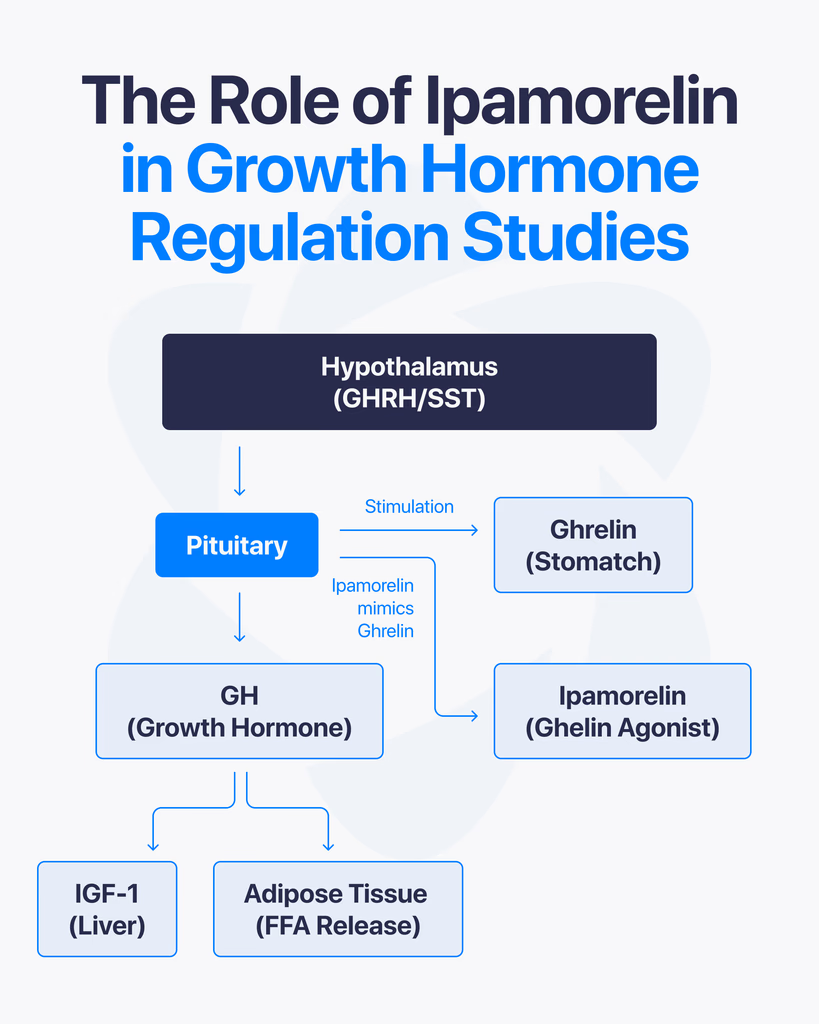
By Dr. Leonard Haberman, Chief Science Officer, OPTMZ Peptides Published March 2, 2026 · Last Updated April 16, 2026 Research Use Only. The information in this article is provided for laboratory research and scientific education. The compounds discussed are not intended for human or animal consumption, clinical use, or therapeutic application. Statements on this site have not been reviewed by the FDA. Ipamorelin is a synthetic pentapeptide studied in endocrine research for its selective stimulation of growth hormone release. Structurally defined as Aib-His-D-2-Nal-D-Phe-Lys-NH₂, it binds the growth hormone secretagogue receptor (GHS-R1a) — the same receptor activated by endogenous ghrelin — and has been characterized in published research as the first GHRP-class ligand with selectivity comparable to GHRH (Raun et al., 1998). This article reviews what the peer-reviewed literature has established about ipamorelin’s mechanism of action, its receptor selectivity, the key preclinical research areas it has been investigated in, and the analytical standards used to verify research-grade ipamorelin purity. What Is Ipamorelin? Ipamorelin is a synthetic five-amino-acid peptide (a pentapeptide) developed in the 1990s as part of research into growth hormone secretagogues — compounds that stimulate endogenous growth hormone (GH) release from the anterior pituitary. It was first characterized by Raun and colleagues at Novo Nordisk in 1998 and described as the first GHRP-receptor agonist whose selectivity for GH release was comparable to that of growth hormone-releasing hormone itself (Raun et al., 1998, PubMed). In current peptide research contexts, ipamorelin is used as a pharmacological tool to investigate GH secretion, GHS-R1a receptor signaling, and related endocrine pathways. It is classified as a research compound and is not approved for clinical therapeutic use in any jurisdiction. What Is the Molecular Structure of Ipamorelin? Ipamorelin’s amino acid sequence is Aib-His-D-2-Nal-D-Phe-Lys-NH₂, where: Aib is α-aminoisobutyric acid, a non-proteinogenic amino acid that introduces conformational rigidity His is L-histidine D-2-Nal is D-2-naphthylalanine, a non-natural D-amino acid D-Phe is D-phenylalanine Lys-NH₂ is C-terminal lysine amide The inclusion of D-amino acids and the unnatural Aib residue is deliberate. These substitutions increase resistance to enzymatic degradation and alter the peptide’s secondary structure, producing a compact, receptor-selective ligand. The molecular formula is C₃₈H₄₉N₉O₅ with a molecular weight of approximately 711.85 g/mol. Structurally, ipamorelin is derived from the GHRP-1 lineage but has been optimized to reduce off-target receptor engagement. This optimization is the basis of the selectivity profile described below. How Does Ipamorelin Stimulate Growth Hormone Release? Ipamorelin is an agonist of the growth hormone secretagogue receptor type 1a (GHS-R1a) — a G-protein-coupled receptor (GPCR) expressed on somatotroph cells of the anterior pituitary and in regions of the hypothalamus. The GHS-R1a is the same receptor that binds endogenous ghrelin. Upon binding, ipamorelin activates the Gq/11 pathway, triggering phospholipase C (PLC) activity, inositol triphosphate (IP₃) generation, and intracellular calcium mobilization. This cascade results in the release of stored growth hormone from somatotroph secretory vesicles into systemic circulation. In pharmacokinetic-pharmacodynamic modeling performed in healthy human volunteers, ipamorelin administration produced a rapid, dose-dependent increase in serum GH, with measurable GH elevation occurring within minutes of subcutaneous or intravenous delivery (Gobburu et al., 1999, PubMed). Unlike synthetic growth hormone itself, which directly supplements circulating GH levels, ipamorelin stimulates the pituitary’s own GH-releasing machinery. Research on GHS-R1a agonists has shown that this mode of action preserves the pulsatile pattern of GH secretion — a pattern considered physiologically important in native endocrine regulation. What Makes Ipamorelin Selective Compared to Other GHRPs? The defining characteristic of ipamorelin in the peer-reviewed literature is its selectivity — specifically, its stimulation of GH release without significant concurrent elevation of adrenocorticotropic hormone (ACTH), cortisol, or prolactin. Earlier growth hormone secretagogues, including GHRP-6 and GHRP-2, reliably stimulate GH release but also produce measurable increases in ACTH and cortisol, and in some studies prolactin as well. This cross-reactivity has historically complicated the use of these earlier peptides as clean research tools for isolating GH-specific effects. Raun et al. (1998) demonstrated in both in vitro and in vivo models that ipamorelin, administered at doses that produced GH responses equivalent to GHRP-6, did not significantly elevate ACTH or cortisol. Johansen et al. (1999) extended these observations and characterized ipamorelin’s receptor binding kinetics, confirming that the selectivity appears to originate from the peptide’s specific binding profile at GHS-R1a and its lack of engagement with receptors responsible for corticotropin release (Johansen et al., 1999, PubMed). For researchers, this matters because it allows the study of GH-dependent pathways with fewer confounding endocrine variables. In bone formation research, for example, Svensson et al. (2000) were able to examine ipamorelin’s influence on bone mineral content in adult rats without simultaneously introducing the catabolic effects of elevated glucocorticoids that would have been expected with less-selective secretagogues (Svensson et al., 2000, PubMed). What Did the Original Discovery Research Establish? Two foundational publications anchor the scientific understanding of ipamorelin. Raun K, Hansen BS, Johansen NL, et al. (1998). Ipamorelin, the first selective growth hormone secretagogue. European Journal of Endocrinology, 139(5):552-561. (PubMed 9849822) — the foundational discovery paper. It established ipamorelin’s potency and efficacy in vitro (using isolated pituitary cells) and in vivo (in swine and rats), benchmarked it against GHRP-6, and documented the distinctive selectivity profile. The paper has been cited more than 180 times in subsequent literature. Johansen PB, Segev Y, Landau D, et al. (1999). Ipamorelin, a new growth-hormone-releasing peptide: Pharmacokinetic and pharmacodynamic properties. (PubMed 10373343) — extended the receptor characterization, established pharmacokinetic parameters in rats, and confirmed in additional models that GH release was not accompanied by ACTH or prolactin elevation. Subsequent research has built on these foundations. Gobburu and colleagues (1999) developed a pharmacokinetic-pharmacodynamic model based on data from human volunteers that described ipamorelin’s GH response curve across a range of doses. The 1998 and 1999 papers remain the primary citations in virtually all contemporary ipamorelin research. How Does Ipamorelin Interact with Somatostatin and Endocrine Feedback? Native GH secretion is regulated by a two-signal system: growth hormone-releasing hormone (GHRH) from the hypothalamus stimulates release, and somatostatin (also hypothalamic) inhibits it. Endogenous ghrelin — and, by extension, GHS-R1a agonists like